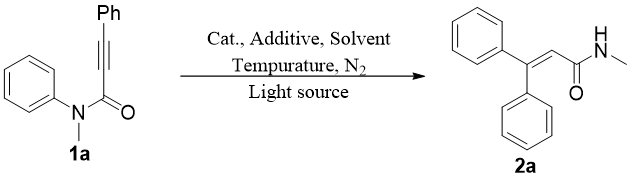
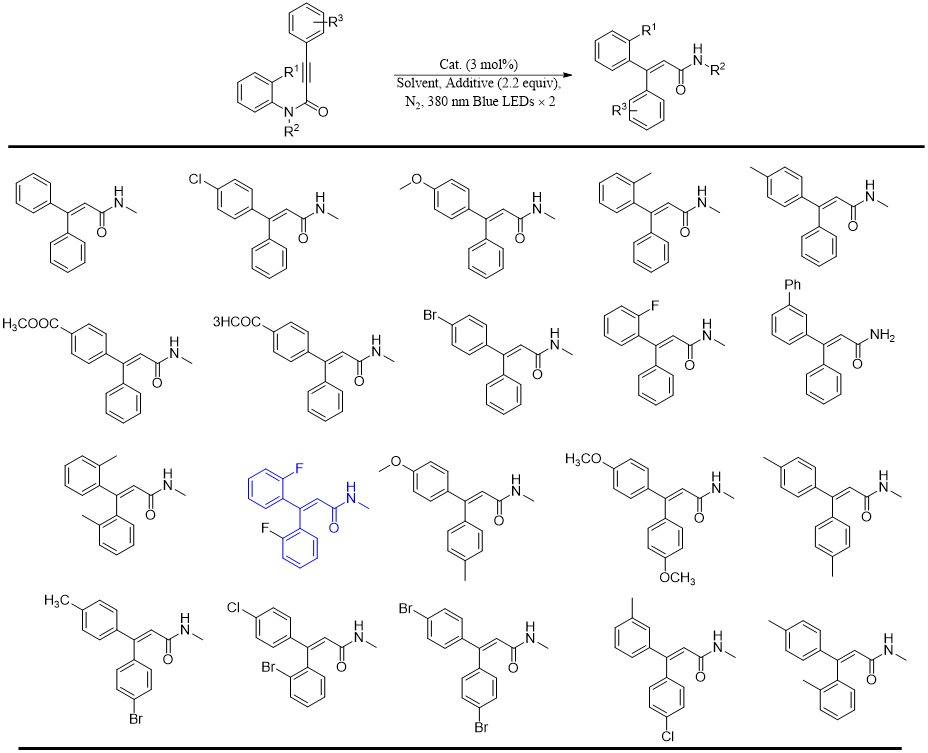
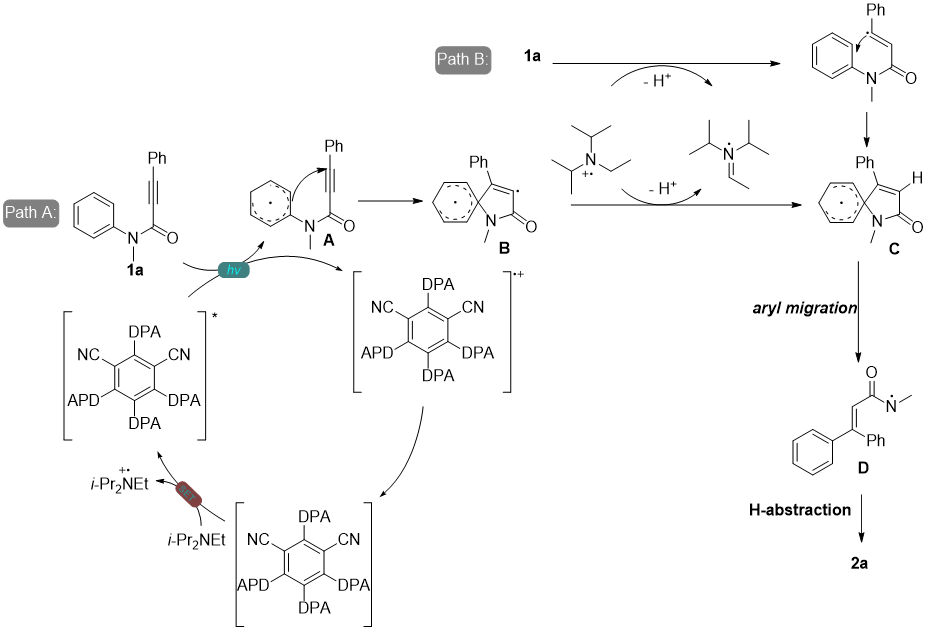
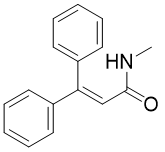
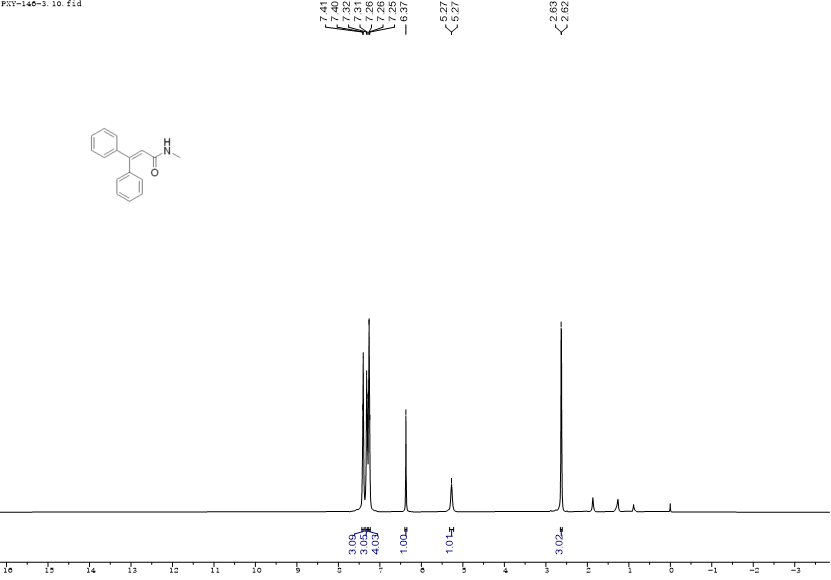
丙烯酰胺,化学式为CR1R2=CHCONR3R4,是一种无色无味的有机小分子,对光线敏感,在紫外线作用下易发生聚合形成聚丙烯酰胺化合物。同时其也是有机合成中的重要基团,存在于蛋白质以及高分子化合物的骨架结构中,因其可以与特定蛋白质的独特半胱氨酸残基发生不可逆的异-迈克尔加成反应形成细胞靶标,使得此类化合物的合成成为药物化学的研究热点[1],因此发展一种简单高效的方法来合成三取代丙烯酰胺具有重要的意义。
传统合成三取代丙烯酰胺的方法中,Wittig-Horner 反应是反应的关键步骤,而它通常产生E/Z异构体的混合物,会根据底物的结构、反应温度、溶剂、催化剂等的变化,E/Z异构体比例也随之变化。在Xiao[2]的合成路线中,化合物5与乙基二乙氧基膦酸乙酸酯的反应主要产生更稳定的顺式中间体9,在顺式形式中,富含电子的羰基氧倾向于向缺电子的吡啶环提供电子,这导致形成更稳定顺式产物6,在经过后续一系列反应后,得到药物分子8。
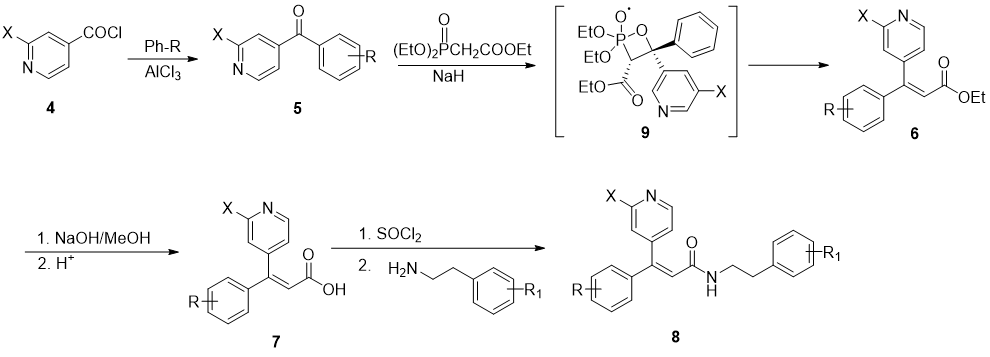
图 6 Wittig-Horner 反应合成丙烯酰胺
乙烯基硅烷是有机化学中一类重要的有价值的组成部分,广泛应用于有机合成(Hiyama-Denmark偶联、Tamao-Fleming氧化等)、高分子化学和药物,但是末端烯烃C(sp2)制备Z-乙烯基硅烷的H键在合成化学中仍然是一个巨大的挑战。张[3]课题组描述了一种高效的钯催化C(sp2)–H硅烷化在温和条件下立体选择性合成Z-乙烯基硅烷,该方法为从简单的起始材料快速合成Z-立体选择性乙烯基硅烷提供了一种实用且环保的策略。他们推测磷酸盐可能促进钯与Pd-Si中间体的解离,以加速催化剂的周转,因此他们采用20 mol%的 (BnO)2PO2H作为磷酸盐,利用环境无害的1,4-苯醌(BQ)作为非金属氧化剂取代化学计量的金属氧化剂,在Pd(OAc)2的催化下合成乙烯基硅烷。该策略为Z-乙烯基硅烷的独家合成提供了区域和立体选择性方案,具有合理到优异的产率和良好的官能团相容性。
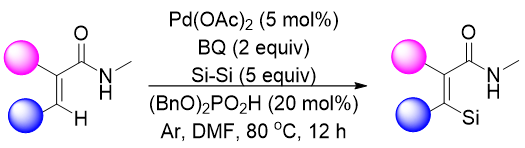
图 7 钯催化合成乙烯基硅烷
2010年,Tsuji[4]首次开发了一种新型钯催化系统,利用钯配合物将甲酰胺分子间加成到炔烃中的方法。甲酰胺与炔烃在钯催化下反应,以酰氯为添加剂,提高(E)-α,β-不饱和酰胺的立体选择性。该催化剂体系实现了在末端炔烃中加入甲酰胺,得到相应的带有末端亚甲基部分的α,β不饱和酰胺作为主要产物,广泛适用于具有各种功能的底物。该方法也可用于N,N-二取代甲酰胺与降冰片烯的反应,以83%产率得到相应产物。
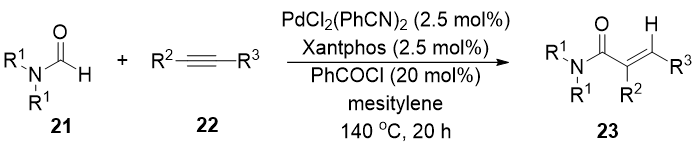
图 8 钯催化合成丙烯酰胺
基于已有的实验数据和前人的相关工作,下图显示了可能的催化循环。通过与 HCl 反应原位生成Pd−H 物质 (A)可能是关键的催化中间体。炔烃(22)与A反应得到烯基Pd中间体(B)(step 1)。然后,可以插入甲酰胺的 C=O 键以形成相应的烷氧基钯中间体 (C)。最后,C继续反应得到所需的产物(23),并且Pd−H物质(A)再生(step3)。或者,可能涉及甲酰基 C−H 键的氧化加成得到 Pd(IV) 中间体 (D) 的循环。从D中还原消除可以提供23,Pd−H物种(A)将再生(cycle II)。
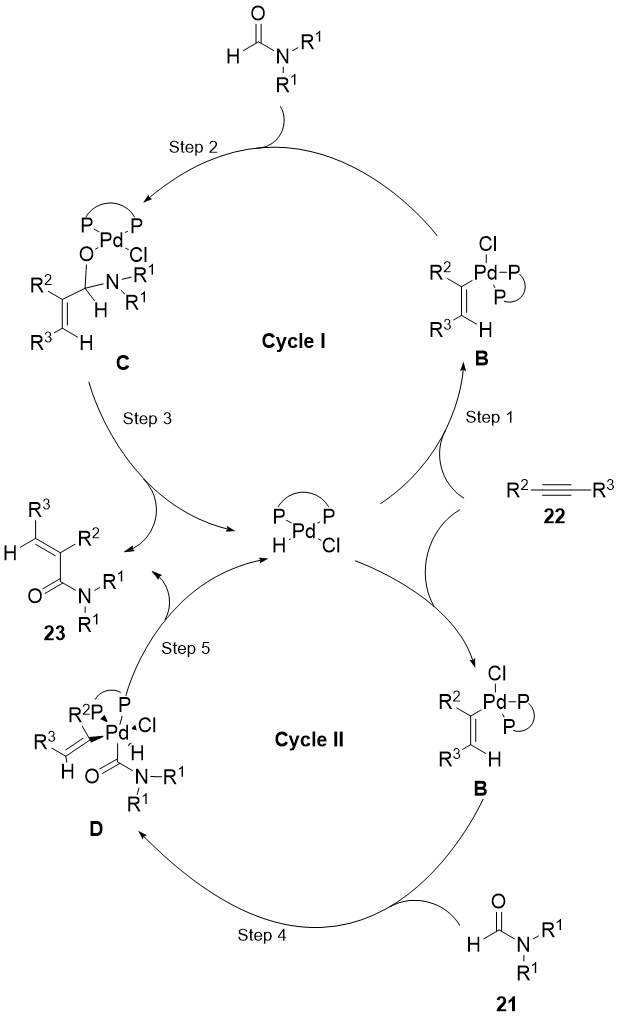
图 9 钯催化合成丙烯酰胺循环
2016年,Ming[5]等人报道了以苯乙烯(化合物24)和胺类化合物(化合物25)为原料,在140 oC的CO气氛下,以CsOAc为碱,3,2-甲氧基苯基做配体,来验证Pd(PPh3)4和Pd(OAc)2对反应的催化效率,结果证明,Pd(OAc)2是最佳催化剂选择,反应延长至48小时,产品的产率提高到70%。但是这种方法中使用的多取代烯基底物几乎与产物一样复杂,这些多取代烯基底物通常不容易有选择地制备,对合成多取代丙烯酰胺具有一定的限制。
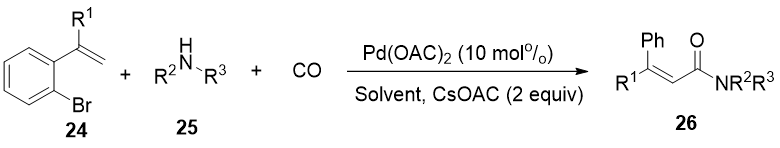
图 10 苯乙烯和胺类反应合成丙烯酰胺
2019年,Russell[6]等人报道了一种制备空气稳定型二氟硼基丙烯酰胺的方法。与普遍存在的有机三氟硼酸盐相比,二氟硼酸丙烯酰胺是非极性的,在交叉偶联反应中,二氟硼基丙烯酰胺底物可提供相应的三取代丙烯酰胺,产率良好。在他们的研究中用(E)-β-硼基丙烯酰胺(化合物27)和KHF2在醋酸中的高效地合成了二氟硼酸丙烯酰胺。接下来,他们将注意力转向交叉偶联反应中二氟硼基丙烯酰胺的反应活性。实验结果表明,在标准的Suzuki - Miyaura交叉偶联条件下,二氟硼基丙烯酰胺可以作为有效的底物与各种富电子和缺电子的芳基溴进行交叉偶联反应,得到相应的三取代丙烯酰胺(化合物29)。
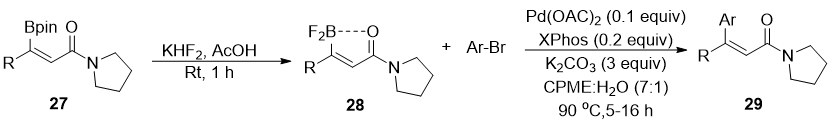
图 11 二氟硼基酰胺交叉偶联合成三取代丙烯酰胺
与各种酮体去饱和方法相比,酰胺的去饱和化更加困难,主要是由于它们的 α-C-H 键的酸度降低。这些去饱和化方法通常依赖于引入α-离去基团,例如卤素、硫、硒衍生物或者TEMPO基团,然后进行消除步骤。虽然这些方法被广泛使用,但需要多个步骤或使用强碱。在此,为实现内酰胺类β-C-C键形成反应目标,Chen[7]等人公开了一种在酸性条件下去饱和合成酰胺的直接方法。这种方法可以在室温下连续操作。该研究以δ-戊丙酰胺作为原料,氧化硫为配体,1,4-二恶烷为溶剂,经过优化后,所需的α,β-不饱和内酰胺通过硼烯醇中间体在室温下以94%的收率获得。这种去饱和方案也可以应用于链状结构,在标准条件下可以得到50%~70%产率的戊唑烷酮衍生的酰胺。
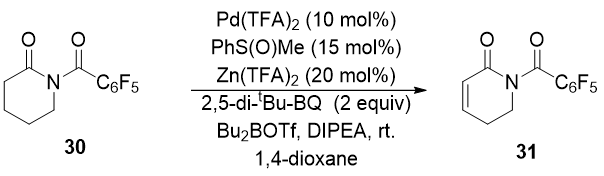
图 12 酸性条件下去饱和N2-保护合成丙烯酰胺
参考文献
[1]冯波, 滕大为, 曹国锐, α,β-不饱和酰胺的合成研究进展,化学通报, 2023, 86, 978-979.
[2]Gopalaiah, K., Kagan, H. B. Use of Nonfunctionalized Enamides and Enecarbamates in Asymmetric Synthesis, Chem. Rev. 2011, 111, 4599-4657.
[3]Courant, T., Dagousset, G. Masson, G.Enamide Derivatives:Versatie Building Blocks for Total Synthesis, Nat. Synth. 2015, 47, 1799-1856.
[4]Song, Z., Lu, T., et al. Stereoselective Simmons-Smith Cyclopropanation of Chiral Enamides. Angew. Chem., Int. Ed. 2007, 46, 4069-4072.
[5]Alix, A., Lalli, C., et al. Highly Enaatioselective Electrophilic α-Bromination of Enecarbamates:Chiral Phosphoric Acid and Calcium Phosphate Salt Catalysts. J. Am. Chem. Soc. 2012, 134, 10389-10392.
[6]Movassaghi, M., Hill, M. D.Synthesis of Substituted Pyridine Derivativas via the Ruthenium-Catalyzed Cycloisomerization of 3-Azadienynes. J. Am. Chem. Soc. 2006, 128, 4592-4593.
[7]Terada, M., Machioka, K., Sorimachi, K.High Substrate/Catalysis by a Chiral Bronsted Acid for an Enantioselective Aza-Ene-Type Reaction. Angew. Chem., Int. Ed. 2006, 45, 2254-2257.
[8]Bai, X. - Y., Wang, Z. - X., et al. Iridium-Catalyzed Enantioselective Hydroalkynylation of Enamides for the Synthesis of Homopropargyl Amides. Angew. Chem., Int. Ed. 2016, 55, 9007-9011.
[9]Daniel, K., Whelligan, Douglas W., et al.Palladium-Catalyzed Intramolecular Amidation of C(sp2)-H Bonds:Synthesis of 4-Aryl-2-quinolinones. J. Org. Chem. 2010, 75, 3900-3903.
[10]Xiao Y. - M., Yang X. - L., et al. Design,Synthesis and Antifungal/Insecticidal Evaluation of Novel Cinnamide Derivatives. Molecules. 2011, 16, 8945-8957.
[11]Wright, S., Petraitis, J., et al. 2,5-Diarylisothiazolone:novel inhibitors of cytokine-induced cartilage destruction. Bioorg. Med. Chem. 1996, 4, 851-858.
[12]Gorsuch, S., Bavetsias, V., et al. Synthesis of isothiazol-3-one derivatives as inhibitors of histone acetytransferases. Bioorg. Med. Chem. 2009, 17, 467- 474.
[13]Chen M. - Y., Xavier P., et al. Access to Isothiazolones from Simple Acrylamides by Pd-Catalazed C-H Bond Activation. Org. Chem. 2019, 84, 13194–13202.
[14]Xiao Y. - M., Yang X. - L., et al.Design,Synthesis and Antifungal/Insecticidal Evaluation of Novel Cinnamide Derivatives. Molecules. 2011, 16, 8945-8957.
[15]Pan J. - L., Chen C., et al. Synthesis of Quinolinones with Palladium-Catalyzed Oxidative Annulation between Acrylamides and Arynes. J. Org. Chem. 2015, 80, 2835-2841.
[16]Markus, W., Upert, G., et al. Palladium-Catalyzed Intermolecular Addition of Formamides to Alkynes. J. Am. Chem. Soc. 2010, 132, 2094-2098.
[17]Li, M., Li, S. - X., Chen, D, - P., et al. Regioselective C-H Active Carbonylation via 1,4-Palladium Migration. Org. Lett. 2023, 25, 2761-2766.
[18]Russell, G., Fritzemeier., Eric, J., et al. Route to Air and Moisture Stable β-Difluoroboryl Acrylamides. Org. Lett. 2019, 21, 8053-8057.
[19]Lies, L., Lsomura, M., et al. Direct Catalytic Desaturation of Lactams Enable by Soft Enolization. J. Am. Chem. Soc. 2017, 139, 7757-7760.
[20]Wu, Y., Zhang, L. - Q., et al. Iridium-Catalyzed Aerobic α,β-Dehydrogenation of Unsaturated Amides and Acids: Activation of Both α- and β- C-H bonds through an Allyl-Iridium Intermediate. J. Am. Chem. Soc. 2018, 140, 735-740.
[21]Chen, M., Dong, G. - B. Copper-Catalyzed Desaturation of Lactones, Lactams, and Ketones under pH-Neutral Conditions. J. Am. Chem. Soc. 2019, 141, 14889-14897.
[22]Duan, X. - L., Zheng, N., et al. Copper-Catalyzed Z-selective Synthesis of Acrylamides and Polyacrylamides via Alkylidene Ketenimines. Nat. Commun., 2022, 13, 4362.
[23]Yuan, Y., Zhao, F. - Q., Wu, X. - F. Copper-catalyzed Enantioselective Carbonylation toward α-Chiral Secondary Amides. Chem. Sci., 2021, 12(38), 12676-12681.
[24]Joanna, D., Nimphius, C., et al. Visible Light Photoredox Catalysis with Transition Metal Complexes: Application in Organic Synthesis. Chem. Rev. 2013, 113, 5322−5363.
[25]Yang, X., Wei, W., Li, H., et al. Oxidative Coupling of Alkenes with Amides using Peroxides: Selective Amide C(sp3)-H Versus C(sp2)-H Functionalization. Chem. Commun. 2014, 50, 12867.
[26]Raymond, J., Cvetovich., et al. Formation of Acrylanilides, Acrylamides, and Amides Directly from Carboxylic Acids Using Thionyl Chloride in Dimethylacetamide in the Absence of Bases. Org. Process Res. Dev. 2006, 10, 944-946.
[27]Ding R. - B., Li Yun., et al. Stereoselective Synthesis of(2Z)-2,4-Dienamides via NBS-Mediated Allyloxyl Addition-Claisen Rearrangement-Dehydrobromination Cascade Reation of Ynsulfonamides. Org. Lett. 2015, 17, 3994–3997.
[28]Santanu, G., Chandan, k., Jana. Aminofluorene-Mediated Biomimetic Domino Amination-Oxygenation of Aldehydes to Amides. Org. Lett. 2016, 18, 5788-5791.
[29]Tebben, L., Prof, D., Studer. Nitroxides: Applications in Synthesis and in Polymer Chemistry. Angew. Chem., Int. Ed. 2011, 50, 5034.
[30]Barham, J. P., Coulthard, G., et al. KOtBU: A Privileged Reagent for Electron Transfer Reactions. J. Am. Chem. Soc. 2016, 138, 7402.
[31]Yang, S., Fan, H. - K., et al. Photoinduced Desaturation of Amides by Palladium Catalysis. Org. Lett. 2022, 24, 6460-6465.
[32]Yu, W. - L., Ren, Z. - G., et al. Cobalt-Catalyzed Chemoselective Dehydrogenation Through Radical Translation Under Visible Light. Chem. Sci., 2022, 13, 7947-7954.
[33]Lu, M. - J., Lin, Z. - W., et al. Visible-Light-Enable Oxidative Coupling of Alkenes with Dialkylformamides To Access Unsaturated Amides. Org. Lett. 2019, 21, 9929-9933.
[34]Luo, J., Zhang, J. Donor-Acceptor Fluorophores for Visible-Light-Promoted Organic Synthesis: Photoredox/Ni Dual Catalytic C(sp3)-C(sp2) Cross-Coupling. ACS Catal., 2016, 6, 873-877.
[35]Singh, A., Singh, K., Weaver, et al. Erratum:Proton Solvation in Protic and Aprotic Solvent. J. Chem., 2015, 776, 51-59.